NeDRexApp: Available Functions
A guide to all the available functions in NeDRexApp.
Import Network from Public Databases
Build and load custom heterogeneous networks from NeDRexDB with different edge types for different purposes via File –> Import
–> Network from Public Databases.... For information about the databases integrated in our knowledgebase see NeDRexDB.
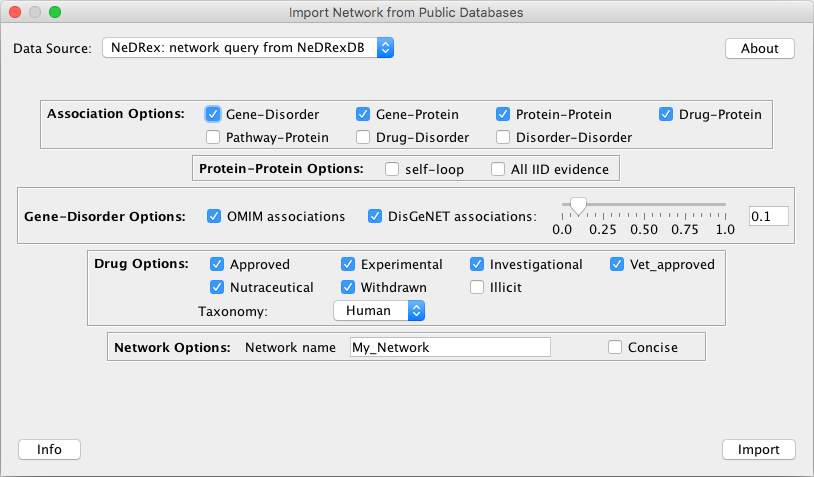
Data Source: NeDRex: network query from NeDRexDB
Association Options: edges with the specified types to be included in the imported graph
Gene-Disorder: Relationships between disorders and genes associated to them
Gene-Protein: Relationships between genes and proteins encoded by them
Protein-Protein: Protein-protein interactions
Drug-Protein: Relationships between drugs and proteins targeted by them
Pathway-Protein: Relationships between proteins and pathways they participate in
Drug-Disorder: Relationships between disorders and the drugs that are indicated for the treatment of disorders
Disorder-Disorder: Relationships representing the disorder hierarchy in MONDO
Protein-Protein Options: Only relevant when the
Protein-Proteinassociation is selected.self-loop: Include self-loop PPIs, by default not added
All IID evidence: Select if you want to include all IID evidence types (experimentally detected, orthologous, computationally predicted). The default experimental option is recommended.
Gene-Disorder Options: Only relevant when the
Gene-Disorderassociation is selected.OMIM associatoins: Check if you want to include disease-gene associations from the OMIM database.
DisGeNET associations: Check if you want to include disease-gene associations from the DisGeNET curated database.
DisGeNET cutoff threshold: For the disease-gene association score. Default: 0 (gives all associations). The DisGeNET score is a measure to rank the gene-disease associations according to their level of evidence.
Drug Options: Only relevant when an association type including drugs is selected.
Drug groups to include. For drug groups definitions refer to the DrugBank website.
Taxonomy: If all selected, non-human drug targets will be included as well, such as bacterial proteins.
- Network Options:
Network name: Enter the name you would like to have assigned to the loaded network
Concise: Check if you want to include a concise list of attributes for nodes and edges in the graph. This does not affect the number of nodes and edges in the network but returns fewer attributes for them to reduce the size of the network file.
It is highly recommended not to select all association types at once and only select those that are necessary for your analyses. You can
find what type of network is required for different types of analysis in the corresponding function in this documentation.
Now you can import your custom network into Cytoscape by clicking on the Import button.
If a network query request with the same selected parameters and association types were submitted before, the network is already built and NeDRexApp just loads that network into Cytoscape. If it’s the first time that such a request is submitted, it takes some time to first build the network in the backend and when the built is finished the app imports the network into Cytoscape.
If you already have some other loaded networks in Cytoscape, when importing a new network from NeDRexDB, it is recommended to always select
--Create new network collection-- option when prompted.
Disease Module Identification
Disease Module Identification can be performed by one of the following algorithms:
BiCoN
DIAMOnD
MuST
Run BiCoN
BiCoN (Biclustering Constrained by Networks) is an unsupervised approach that conducts simultaneous patient and gene clustering such that the genes that provide the best possible clustering are also connected in the PPI network (Lazareva et al. 2021). For more information regarding the theoretical background of BiCoN algorithm, refer to the algorithm section (BiCoN).
Starting point:
A gene expression, methylation or any other numerical data file for patients, to be uploaded by the user. The numerical data is accepted in the following format as a tabular file (.csv, .tsv, or .txt):
genes as row
patients as columns
first column - genes IDs (Entrez IDs)
For instance:
GeneID |
Patient 1 |
Patient 2 |
Patient 3 |
Patient 4 |
|---|---|---|---|---|
6311 |
7.3487 |
6.9464 |
7.2675 |
6.8363 |
133584 |
6.5562 |
5.7931 |
6.3360 |
5.6389 |
283165 |
3.8878 |
3.9588 |
4.0173 |
4.0923 |
You can download an example file for lung cancer here.
It contains a Non-Small Cell Lung Cancer dataset from GEO (GSE30219) for patients with either adenocarcinoma or squamous cell carcinoma.
Options:
- Algorithm settings:
Minimal solution subnetwork size: The lower bound for size of the solution subnetwork
Maximal solution subnetwork size: The upper bound for size of the solution subnetwork
Input file for numerical patient data: Input file should contain gene expression, methylation or any other kind of numerical data for patients in the format specified above.
- Result network:
Use custom name for the result network: Select if you would like to use your own custom name for the result network, otherwise a default name based on the selected algorithm parameters will be assigned
Name of the result network: Enter the name you would like to have assigned to the result network
- Returns:
A connected subnetwork of genes which is the union of two identified gene clusters (red and blue)
A downloadable heatmap of biclustering result (in the Results Panel)
A downloadable table with the patients’ groups assignments (in the Results Panel)
The resulting subnetwork is colored with respect to its difference in expression patterns in patients clusters. In the Results panel a
clustermap of the achieved biclustering solution along with the known patients’ groups can be found. If you run multiple analyses with different
parameters, the images and tables will be appended in the Results Panel.
To see an example of how this function can be used, please see our HD use case with BiCoN or the section BiCoN in typical steps for a more general explanation.
Run DIAMOnD
The DIAMOnD (DIseAse MOdule Detection) algorithm identifies the disease module around a set of known disease genes/proteins (seeds) based on the principle that the connectivity significance is highly distinctive for known disease proteins (Ghiassian et al. 2015). For more information regarding the theoretical background of DIAMOnD, refer to the algorithm section (DIAMOnD).
Starting point:
A set of genes/proteins (seeds), either from
the result of Get Disease Genes function or
a custom list either supplied by:
the current selection in the network (e.g. obtained by
Select nodes–>All nodes of specific type) oruploading a file containing a list of e.g. differentially expressed genes (see
Input seedsoption)
Options:
- Algorithm settings:
Number of DIAMOnD genes (iteration): desired number of DIAMOnD genes/proteins, or number of iterations.
Weight of seeds: Alpha parameter, representing the initial weight of the seeds, default value: 1.
- Input seeds:
Read seeds from a file: If selected, gene seeds will be read from a file. Otherwise, the selected nodes in the current network will be taken as seeds.
Input file for seeds: Input file containing a list of seeds, one seed per line. The file should be a tab-separated file and the first column will be taken as seeds. Genes with Entrez ID are acceptable as input.
- Result network:
Return all edges in the result disease module: If selected, all edges between genes in the result disease module will be returned, otherwise only edges between seeds and new nodes will be returned. For large disease modules, the latter option is recommended for better visualization.
Use custom name for the result network: Select, if you would like to use your own name for the result network, otherwise a default name based on the selected algorithm parameters will be assigned.
Name of the result network: Enter the name you would like to have assigned to the result network.
Returns:
A new network containing the starting seeds and the genes found by DIAMOnD.
The rank of DIAMOnD nodes in the disease module is returned as node attribute (rank) in the node table of the created network.
To see examples of how this function can be used, please see our PE use case with DIAMOnD and AD use case with hyperlipidemia as original indication. For a more general explanation see the section DIAMOnD in typical steps .
Run MuST
By selecting proteins associated with a disease under study (seeds), MuST (Multi-Steiner trees) extracts a connected subnetwork involved in the disease pathways based on the aggregation of several non-unique approximates of Steiner trees (Sadegh et al. 2020). For more information regarding the theoretical background of MuST, refer to the algorithm section (MuST).
Starting point:
A set of genes/proteins (seeds), either from
the result of Get Disease Genes function or
a custom list either supplied by:
the current selection in the network (e.g. obtained by
Select nodes–>All nodes of specific type) oruploading a file containing a list of e.g. differentially expressed genes (see
Input seedsoption)
Options:
- Algorithm settings:
Return multiple Steiner trees: the solution to the Steiner tree problem is usually non-unique. It’s recommended to choose this option to get aggregation of several results.
The number of Steiner trees: the number of Steiner trees to be returned.
Max number of iterations: the maximum number of iterations that the algorithm runs to find dissimilar Steiner trees of the selected number.
Penalize hub nodes: Penalize high degree nodes by incorporating the degree of neighboring nodes as edge weights.
Hub penalty: Penalty parameter for hubs. Sets edge weight to (1 - hub_penalty) * AveDeg(G) + (hub_penalty / 2) * (Deg(source) + Deg(target))
- Input seeds:
Read seeds from a file: If selected, gene seeds will be read from a file. Otherwise, the selected nodes in the current network will be taken as seeds.
Input file for seeds: Input file containing a list of seeds, one seed per line. The file should be a tab-separated file and the first column will be taken as seeds. Genes with Entrez ID are acceptable as input.
- Result network:
Use custom name for the result network: Select, if you would like to use your own name for the result network, otherwise a default name based on the selected algorithm parameters will be assigned.
Name of the result network: Enter the name you would like to have assigned to the result network.
Returns:
A new network containing all trees found by MuST, including the starting seeds and the connector nodes.
The number of different Steiner trees nodes/edges participate in are returned as node/edge attributes (participation_number) in the node/edge table of the created network.
To see examples of how this function can be used, please see our OC use case with MuST and IBD use case with MuST. For a more general explanation see the section MuST in typical steps .
Drug Prioritization
Drugs targeting directly or the vicinity of the selected genes/proteins (seeds) can be ranked by one of the following algorithms:
TrustRank
Closeness centrality
Drug prioritization can be performed either directly on the genes associated with diseases obtained by Get Disease Genes function, or on the entire disease module obtained by Disease Module Identification, or on any selected set of genes/proteins (seeds) compiled by your expert knowledge.
All drugs targeting the selection
This function does not rank the drugs but returns all drugs targeting the selection.
Starting point:
A set of genes/proteins (seeds), either
selection of all or some of genes/proteins in the returned disease module or
selection of disease genes returned by Get Disease Genes function.
Options:
- Drug option:
Include only approved drugs: Specifies, whether only approved drugs targeting the selection should be considered or all drugs.
- Result network:
Use custom name for the result network: Select, if you would like to use your own name for the result network, otherwise a default name will be assigned.
Name of the result network: Enter the name you would like to have assigned to the result network.
Returns: A new network containing the seeds used as input together with drugs targeting them.
See our PE use case and AD use case with diabetes as original indication for examples of how this function can be used.
Rank drugs with Closeness Centrality
Closeness is a node centrality measure that ranks the nodes in a network based on the lengths of their shortest paths to all other nodes in the network. Here, we implemented a modified version, where closeness is calculated with respect to only the selected seeds. For more information regarding the theoretical background of Closeness Centrality, refer to the algorithm section (Closeness centrality).
Starting point:
A set of genes/proteins (seeds), either
selection of all or some of genes/proteins in the returned disease module or
selection of disease genes returned by Get Disease Genes function.
Options:
- Closeness centrality algorithm settings:
Include only direct drugs: Specifies, whether only drugs targeting seeds should be considered or all drugs.
Include only approved drugs: Specifies, whether only approved drugs targeting seeds should be considered or all drugs.
The number of top-ranked drugs: Specifies the number of top-ranked drugs to be returned in the result network.
- Result network:
Use custom name for the result network: Select, if you would like to use your own name for the result network, otherwise a default name based on the selected algorithm parameters will be assigned.
Name of the result network: Enter the name you would like to have assigned to the result network.
Returns:
A new network containing the seeds used as input together with top-ranked drugs associated to the seeds.
The rank of drugs is returned as node attribute (rank) in the node table of the created network.
See our AD use case with hypertension as original indication, AD use case with hyperlipidemia as original indication and IBD use case for examples of how this function can be used. For a more general explanation, see the section Closeness centrality in typical steps.
Rank drugs with TrustRank
TrustRank ranks nodes in a network based on how well they are connected to a (trusted) set of seed nodes. It is a modification of Google’s PageRank algorithm, where “trust” is iteratively propagated from seed nodes to adjacent nodes using the network topology. For more information regarding the theoretical background of TrustRank, refer to the algorithm section (TrustRank).
Starting point:
A set of genes/proteins (seeds), either
selection of all or some of genes/proteins in the returned disease module or
selection of disease genes returned by Get Disease Genes function.
Options:
- TrustRank algorithm settings:
Include only direct drugs: Specifies, whether only drugs targeting seeds should be considered or all drugs.
Include only approved drugs: Specifies, whether only approved drugs targeting seeds should be considered or all drugs.
Damping factor: The larger the damping factor, the faster the trust is propagated through the network. Default: 0.85.
The number of top-ranked drugs: Specifies the number of top-ranked drugs to be returned in the result network.
- Result network:
Use custom name for the result network: Select, if you would like to use your own name for the result network, otherwise a default name based on the selected algorithm parameters will be assigned
Name of the result network: Enter the name you would like to have assigned to the result network
Returns:
A new network containing the seeds used as input together with top-ranked drugs associated to the seeds.
The rank of drugs is returned as node attribute (rank) in the node table of the created network.
See our PE use case and HD use case for examples of how this function can be used. For a more general explanation, see the section TrustRank in typical steps.
Exploratory Functions
Get drugs indicated in disease
Required imported network from NeDRexDB:
A network with at least Drug-Disorder association type.
Starting point: A set of selected disease nodes in the network.
Options:
Include subtypes of selected disorders: Specifies whether only the selected disorders should be considered or also their first level subtypes (based on disease hierarchy from MONDO).
Include all subtypes of disorders: If selected, all the subtypes of the selected disorders (all descendants in the disease hierarchy from MONDO and not just the first level of subtypes) will be considered.
Returns: A network containing the selected disorders (and their subtypes) and all drugs indicated for treatment of them.
Neighbor Module from Seed Proteins
Required imported network from NeDRexDB:
A network with at least Protein-Protein association type. For full function, a network with Gene-Disorder, Gene-Protein and Protein-Protein association types.
Starting point: A selection of protein nodes in the network.
Options:
- Edge types to include in the module:
Protein-Protein: Include first neighboring Protein-Protein interactions
Protein/Gene-Disorder: Include disorders associated with proteins/genes
Collapse proteins on genes: Collapse proteins on their encoding genes in the created subnetwork
- Result network:
Use custom name for the result network: Select, if you would like to use your own name for the result network, otherwise a default name will be assigned.
Name of the result network: Enter the name you would like to have assigned to the result network.
Returns:
Creates a subnetwork including the direct neighborhood of the primary selected proteins along with their Subnetwork Participation Degree as node attribute (subnet_participation_degree).
There’s also an option to include the diseases associated with these proteins.
Subnetwork Participation Degree (SPD) is the degree of the protein node within the subnetwork, normalized by the degree of the node in the full PPI network. The SPD quantifies how enriched the interactions of a given protein are in a given subnetwork.
Start with Disease -> Drugs
Required imported network from NeDRexDB:
A network with at least Gene-Disorder, Gene-Protein, Protein-Protein and Drug-Protein association types.
Starting point: A selection of disease node(s) in the network.
Options:
Include Steiner tree in analysis: Checking the box includes the additional proteins found by Steiner tree connecting proteins associated with the given set of disorders in the analysis.
Collapse proteins on their encoding genes: Collapse proteins on their encoding genes in the created subnetwork
- Result network:
Use custom name for the result network: Select, if you would like to use your own name for the result network, otherwise a default name will be assigned.
Name of the result network: Enter the name you would like to have assigned to the result network.
Returns: Starting with a set of selected diseases, a subnetwork containing Disease->Gene->Protein->Drug paths will be returned. There’s an option to run Steiner tree on the intermediate genes/proteins to expand the exploration.
Start with Drug -> Diseases
Required imported network from NeDRexDB:
A network with at least Gene-Disorder, Gene-Protein, Protein-Protein and Drug-Protein association types.
Starting point: A selection of drug node(s) in the network.
Options:
Include Steiner tree in analysis: Checking the box includes the additional proteins found by Steiner tree connecting proteins targeted by the set of drugs in the analysis.
Collapse proteins on their encoding genes: Collapse proteins on their encoding genes in the created subnetwork
- Result network:
Use custom name for the result network: Select, if you would like to use your own name for the result network, otherwise a default name will be assigned.
Name of the result network: Enter the name you would like to have assigned to the result network.
Returns: Starting with a set of selected diseases, a subnetwork containing Drug->Protein->Gene->Disease paths will be returned. There’s an option to run Steiner tree on the intermediate genes/proteins to expand the exploration.
Projections of Heterogeneous Network
Diseasome
Required imported network from NeDRexDB:
For the projected network based on shared drugs, a network with at least Drug-Disorder association type.
For the projected network based on shared genes, a network with at least Gene-Disorder association type.
Options:
- Create Diseasome based on:
shared drugs
shared genes
Returns: Creates a Disease-Disease projection of the heterogeneous network. If shared drugs was selected, there is an edge between a pair of diseases when they have at least one drug indicated for treatment of them in common. If shared genes was selected, there is an edge between a pair of diseases when they have at least one gene associated to them in common. The number of shared drugs/genes and jaccard index are returned as edge attributes in the edge table of the created network.
Drugome
Required imported network from NeDRexDB:
For the projected network based on shared indication, a network with at least Drug-Disorder association type.
For the projected network based on shared targets, a network with at least Drug-Protein association type.
Options:
- Create Drugome based on:
shared indications
shared targets
Returns: Creates a Drug-Drug projection of the heterogeneous network. If shared indication was selected, there is an edge between a pair of drugs when they have at least one indication in common. If shared targets was selected, there is an edge between a pair of drugs when they have at least one target in common. The number of shared indications/targets and jaccard index are returned as edge attributes in the edge table of the created network.
Quick Start with Drug Repurposing
To quickly find potential repurposable drugs for the selected disease, with one click, Quick Start performs all steps of the drug repurposing workflow:
Get disease associated genes
Run disease module identification
Run drug prioritization
Required imported network from NeDRexDB:
A network containing at least Gene-Disorder and Disorder-Disorder association types.
Starting point: Selected disorder(s) in the network.
Method selection: User can select which disease module identification methods to be applied.
MuST: with preset parameters #trees=5, max-iter=5, hp=0, for more information see MuST function.
DIAMOnD: with preset parameters iter=200, weight (alpha)=1, for more information see DIAMOnD function.
- Result network:
Use custom name for the result network: Select, if you would like to use your own name for the result network, otherwise a default name based on the selected algorithm parameters will be assigned.
Name of the result network: Enter the name you would like to have assigned to the result network.
Returns: The identified disease module together with 100 top-ranked drugs targeting the module.
All genes associated to the selected disorders (and their subtypes) will be considered as seeds for the module identification step. Then all the genes in the returned disease module will be considered as seeds for drug ranking with Closeness centrality (including only approved and direct drugs).
Quick Select
This function helps to quickly select the nodes in the loaded network. The nodes can be found by partially matching the query typed into the respective field.
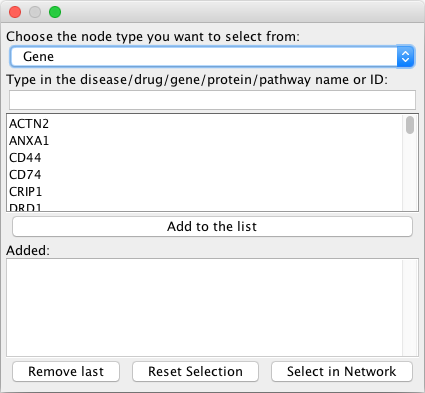
Options:
Choose the node type you want to select from: Select whether the query is going to match a disorder, drug, gene, protein or pathway.
Type in the disease/drug/gene/protein/pathway name or ID: This is the query. You can either type in the name of the node you are looking for (from the field
displayName), the ID (e.g. Entrez ID for genes, Uniprot AC for proteins, MONDO ID for diseases, DrugBank ID for drugs and Reactome ID for pathways) or for proteins the gene name.
After having selected an entry, it is added to the list by clicking on the Add to the list button. After having selected all nodes that
you are interested in, click on Select in Network. If something was added by mistake, the last element can be removed
from the list with Remove last, the selection in the graph can be reset with Reset Selection.
Returns:
All elements in the list are selected in the current network after clicking on Select in Network.
For examples of how to use this function, please refer to the Section Use cases.
Get Disease Genes
Required imported network from NeDRexDB:
A network containing at least Gene-Disorder and Disorder-Disorder association types. Based on the user selection of databases (OMIM, DisGeNET)
and cutoff threshold at importing step, the result of this function can vary.
Starting point: A set of selected disease nodes in the network.
Options:
Include all subtypes of disorders: If selected, all the subtypes of selected disorders (all descendants in the disease hierarchy from MONDO) will be considered. Otherwise, only the selected disorders will be considered.
- Export to file:
Export the obtained disease genes to a file: If selected, you can export the obtained disease genes to a file for later usage. Since the Gene-Disorder associations from OMIM database are updated weekly, we recommend to use the
Export to fileoption in order to be able to reproduce your results later.Select the path to output file: Specify the path where the file should be saved and how it should be named.
- Result network:
Use custom name for the result network: Select, if you would like to use your own name for the result network, otherwise a default name will be assigned.
Name of the result network: Enter the name you would like to have assigned to the result network.
Returns: A network containing the selected disorders (and their subtypes) and all genes associated to them.
For a use case of this function, see Option 1 of Section Step 1: Select disease and associated genes or Section Use cases.
Select nodes
Various functions to select nodes in the network.
From file
Starting point: A file containing a list of IDs (one ID per line) to be selected in the current network. The IDs will be matched to the specified column in the node table (node attribute) of the current network.
Options:
Select nodes with exact matching name: If the box is not checked, the result will also include the nodes whose names partly match with the given list.
The column to select node IDs from: The node attribute (column in the node table) to which the list of IDs (partly) will be matched.
File path: Path to the file containing the list.
Returns: In the current network selects nodes that (partly) matches the provided list of IDs.
For a use case of this function, see Option 2 of Section Step 1: Select disease and associated genes.
All nodes of specific type
Starting point: The current network should contain type as node attribute (column in the node table).
Options:
Node type: Select all nodes of this type in the current network.
Returns: In the current network selects all nodes of the specified type.
To see examples of how this function can be used, please see our OC use case or IBD use case.
Neighbors of specific type
Starting point: A set of selected nodes in the current network and the network should contain type as node attribute (column in the node table).
Options:
Neighbor type: Select all nodes of this type which are direct neighbors of selected nodes.
- Edge selection:
Only edges connecting selected nodes and the neighbors of the specified type
All edges between nodes (selected + neighbors of specified type)
- New network:
Create a new network from selection: If selected, it also creates a new network from the selected nodes and edges.
Use custom name for the new network: Select, if you would like to use your own name for the new network, otherwise a default name will be assigned.
Name of the new network: Choose how you want to name the new network.
Returns: In the current network, it selects those direct neighbors of selected nodes matching the specified node type. If opted for, additionally it creates a new network from the aforementioned selection.
Supplementary Functions
Induced subnetwork of selected nodes
Starting point: A selection of nodes in the network.
Options:
Create a new network from the induced network: If selected, a new network from the induced network of the selected nodes is created. Otherwise, the edges of the induced subnetwork will just be selected in the current network.
- Result network:
Use custom name for the induced network: Select, if you would like to use your own name for the result network, otherwise a default name will be assigned
Name of the induced network: Enter the name you would like to have assigned to the result network
Returns: The induced subnetwork, which is formed from the selected nodes in the network and all of the edges connecting node pairs in that subset.
Map selection to another network
Starting point: A selection of nodes and/or edges in the network.
Options:
Name of the target network to map the selection to: Specify to which of the existing networks should the selection be mapped.
Returns: The selected nodes and edges in the current network will be selected in the specified target network if they exist there.
Hint: This function is specifically useful when you want to continue exploring the nodes from your current network/analysis in another network.
For example, you obtained a disease module by one of the Disease Module Identification methods and now you are interested to find to what other diseases
the genes from your module are associated with. For this purpose, you need to import a network containing Gene-Disorder associations from NeDRexDB as explained
in Import Network from Public Databases and then map the genes from your disease module to this imported network by Map selection to another network. After mapping the nodes to the imported network,
you need to select that network and via NeDRex –> Select Nodes –> Neighbors of specific types and selecting Disorder as neighbor type obtain the disorders.
Validation
To validate the statistical significance of the lists of drugs and disease modules returned by NeDRex, three main validation methods are implemented in the App, each with two variations. As reference, all approaches require a list of drugs indicated for the treatment of the disease of interest. This list can either be provided by the user or be obtained directly from NeDRexDB. For more information regarding the statistical validation approach, refer to the algorithm section (Statistical Validations).
Drug List
Starting point: The result subnetwork from a performed Drug Prioritization task. Alternatively, a file containing a list of drugs (together with their ranks) to be statistically validated. Furthermore, a file containing a list of known drugs indicated for the treatment of disease or in clinical trials for the disease should be available.
Options:
- Algorithm settings:
Number of permutations: The number of random lists of drugs to build background distribution. The default 1000 is a reasonable choice.
Include only approved drugs: If selected, only approved drugs, registered based on DrugBank database, will be taken into account for the statistical validation
- Reference drugs:
Read drugs indicated for disease from a file: If selected, drugs indicated for treatment of disease will be read from a file. Otherwise, selected drugs in the current network will be taken as indicated drugs. For more information about how to create such a list refer to the algorithm section (Statistical Validations).
Input file for drugs indicated for disease: Input file containing list of drugs indicated for treatment of disease, one drug per line. The file should be a tab-separated file and the first column will be taken as drugs. Drugs with DrugBank IDs are acceptable as input.
- Drugs for validation
Read drugs to be validated from a file: If selected, a list of drugs to-be-validated will be read from a file. Otherwise, all drugs in the current network with their ranks from node table will be taken as drugs to-be-validated.
Input file for drugs to be validated: Input file containing list of drugs with their ranks, one drug per line. The file should be a tab-separated file, the first column will be taken as drugs and second column as ranks. Drugs with DrugBank IDs are acceptable as input.
- Validation result:
Description of the validation run: Write in the description of the validation job you will be running to be shown in the result panel. For example, name of the disease. This helps tracking your analyses later.
Returns: Two variations of the empirical P-value in the Results Panel.
Disease Module
Starting point: The result subnetwork from a performed Disease Module Identification task. Alternatively, a file containing the list of the disease module’s genes/proteins to be statistically validated. Furthermore, a file containing a list of known drugs indicated for the treatment of disease or in clinical trials for the disease should be available.
Options:
- Algorithm settings:
Number of permutations: The number of random lists of drugs to build background distribution. The default 1000 is a reasonable choice.
Include only approved drugs: If selected, only approved drugs, registered based on DrugBank database, will be taken into account for the statistical validation
- Reference drugs:
Read drugs indicated for disease from a file: If selected, drugs indicated for treatment of disease will be read from a file. Otherwise, selected drugs in the current network will be taken as indicated drugs. For more information about how to create such a list refer to the algorithm section (Statistical Validations).
Input file for drugs indicated for disease: Input file containing list of drugs indicated for treatment of disease, one drug per line. The file should be a tab-separated file and the first column will be taken as drugs. Drugs with DrugBank IDs are acceptable as input.
- Disease module
Read disease module from a file: If selected, disease module’s genes/proteins will be read from a file. Otherwise, all the genes/protein in the current network will be taken as disease module.
Input file for disease module: Input file containing list of module’s genes/proteins, one entity per line. The file should be a tab-separated file, the first column will be taken as module’s gene/protein. Entrez IDs for genes and Uniprot AC for proteins are acceptable.
- Validation result:
Description of the validation run: Write in the description of the validation job you will be running to be shown in the result panel. For example, name of the disease. This helps tracking your analyses later.
Returns: Two variations of the empirical P-value in the Results Panel.
Joint Module & Drugs
Starting point: The result subnetwork after running one of the Disease Module Identification functions and one of the Drug Prioritization functions in a sequence. Alternatively, a file containing a list of drugs to be statistically validated, together with a file containing the disease module’s genes/proteins. Furthermore, a file containing a list of known drugs indicated for the treatment of disease or in clinical trials for the disease should be available.
Options:
- Algorithm settings:
Number of permutations: The number of random lists of drugs to build background distribution. The default 1000 is a reasonable choice.
Include only approved drugs: If selected, only approved drugs, registered based on DrugBank database, will be taken into account for the statistical validation
- Reference drugs:
Read drugs indicated for disease from a file: If selected, drugs indicated for treatment of disease will be read from a file. Otherwise, selected drugs in the current network will be taken as indicated drugs. For more information about how to create such a list refer to the algorithm section (Statistical Validations).
Input file for drugs indicated for disease: Input file containing list of drugs indicated for treatment of disease, one drug per line. The file should be a tab-separated file and the first column will be taken as drugs. Drugs with DrugBank IDs are acceptable as input.
- Drugs for validation
Read drugs to be validated from a file: gIf selected, a list of drugs to-be-validated will be read from a file. Otherwise, all drugs in the current network will be taken as drugs to-be-validated.
Input file for drugs to be validated: Input file containing list of drugs with their ranks, one drug per line. The file should be a tab-separated file, the first column will be taken as drugs. DrugBank IDs are acceptable.
- Disease module
Read disease module from a file: If selected, disease module’s genes/proteins will be read from a file. Otherwise, all the genes/protein in the current network will be taken as disease module.
Input file for disease module: Input file containing list of module’s genes/proteins, one entity per line. The file should be a tab-separated file, the first column will be taken as module’s gene/protein. Entrez IDs for genes and Uniprot AC for proteins are acceptable.
- Validation result:
Description of the validation run: Write in the description of the validation job you will be running to be shown in the result panel. For example, name of the disease. This helps tracking your analyses later.
Returns: Two variations of the empirical P-value in the Results Panel.
MuST on current network
This function is similar to MuST from Disease Module Identification but can be run on any custom PPI network loaded in Cytoscape. For more about the theory behind MuST, see the algorithms section MuST.
Starting point: A selection of proteins / genes (seeds) in the current network.
Options:
- Algorithm settings:
Return multiple Steiner trees: the solutions to the Steiner tree problem are usually non-unique and computing several Steiner trees increases the stability of the extracted mechanism.
The number of Steiner trees: the number of Steiner trees to be returned.
Max number of iterations: the maximum number of iterations that the algorithm runs to find dissimilar Steiner trees of the selected number.
Penalize hub nodes: Penalize high degree nodes by incorporating the degree of neighboring nodes as edge weights.
Hub penalty: Penalty parameter for hubs. Sets edge weight to (1 - hub_penalty) * AveDeg(G) + (hub_penalty / 2) * (Deg(source) + Deg(target))
- Result network:
Use custom name for the result network: Select, if you would like to use your own name for the result network, otherwise a default name based on the selected algorithm parameters will be assigned
Name of the result network: Enter the name you would like to have assigned to the result network
Returns:
A new network containing all trees found by MuST, including the starting seeds and the connector nodes.
The number of different Steiner trees nodes participate in are returned as node attribute (#participation) in the node table of the created network.
Create NeDRex visual style
NeDRex visual style is specifically suitable for networks generated by NeDRexAPP or imported from NeDRexDB.
However, it can also be applied to any network with a type column that contains at least one of the types Disorder, Drug, Gene, Pathway, Protein.
The attribute displayName from node table is passed for the node label.
By running this function a visual style with the following format will be created:
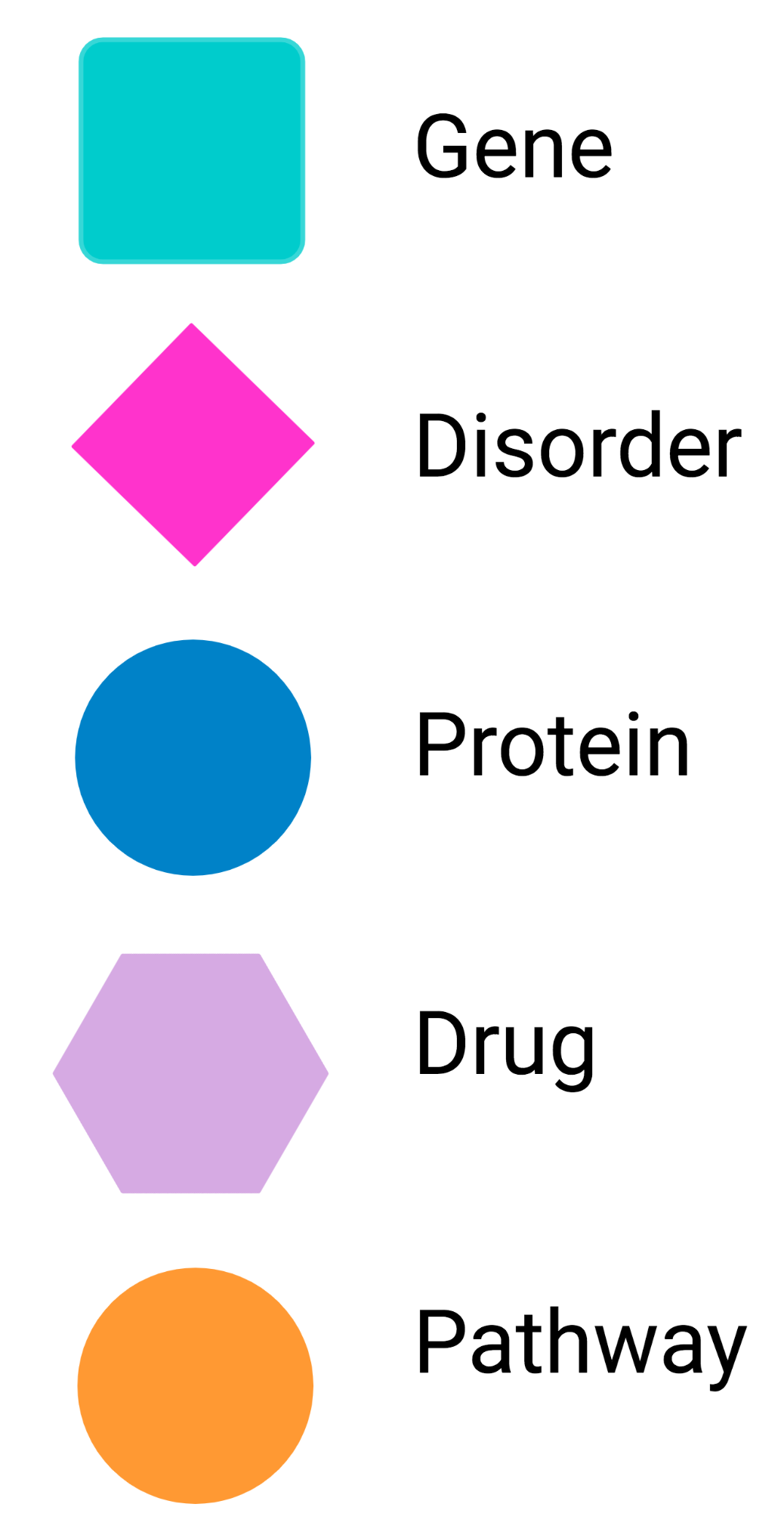
The NeDRex visual style is then added to the Styles available in Cytoscape. This visual style will be automatically applied to the newly created
networks by NeDRexAPP functions. You can also apply the style to any network created before by selecting the NeDRex style from the available styles in the corresponding tab.
Show NeDRex Results Panel
Opens the NeDRex Results Panel. The results from previously run tasks of some functions (e.g. BiCoN or Validation) will be shown in this panel.
The NeDRex Results Panel also appears automatically after running the aforementioned tasks. Each individual result can also be deleted from this panel.
If BiCoN has been run, the result panel should look something like the following:
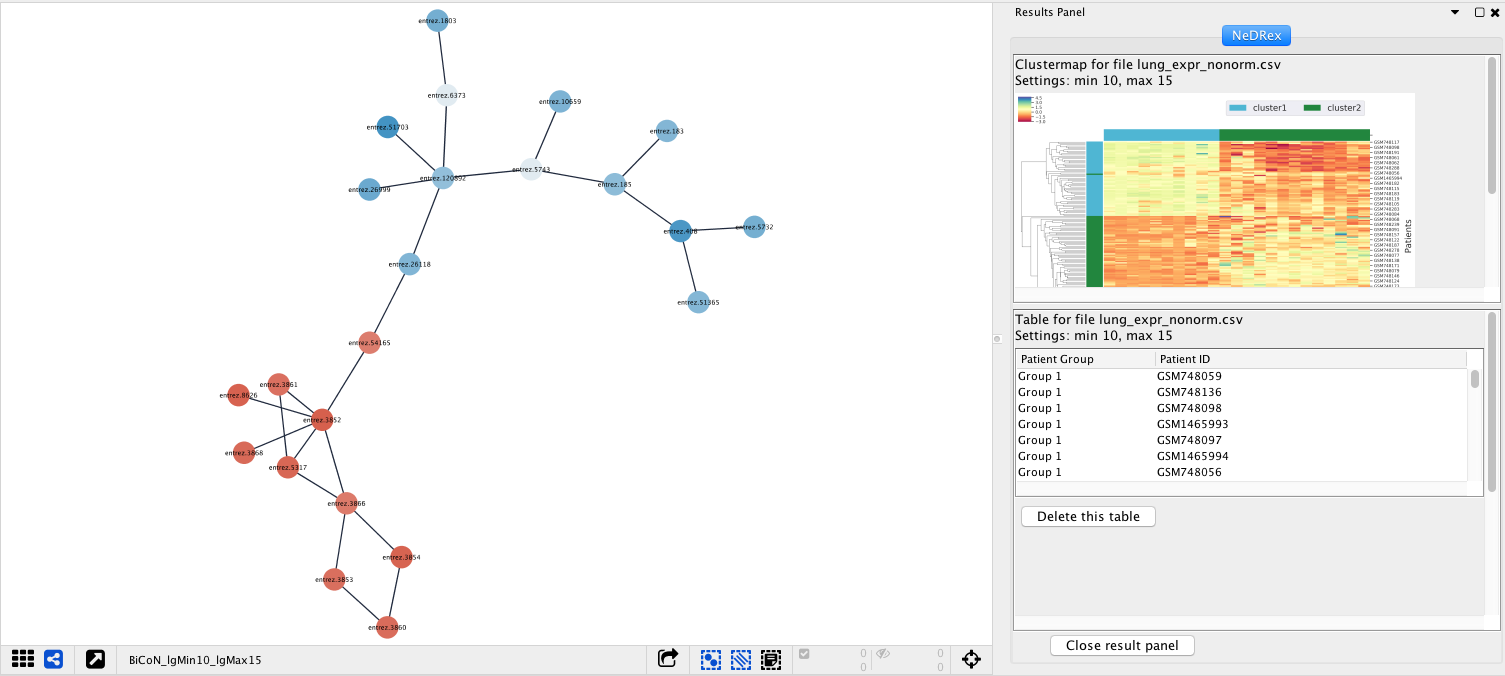
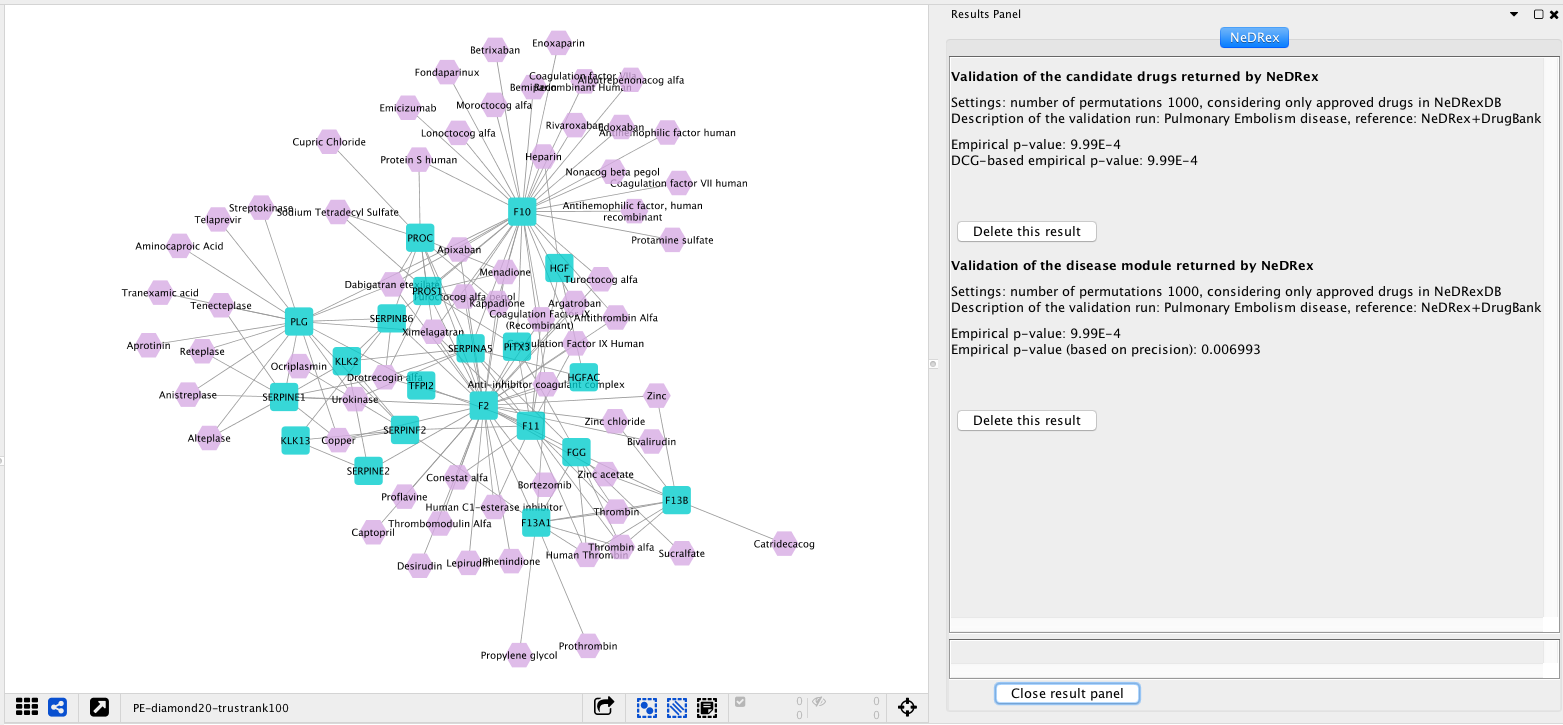
After running a Validation job, the result panel should look something like the following:
Right Click Functions
The following two functions are available via right-click context menu of nodes.
Open entries in the database
By right clicking on a node of interest and choosing NeDRex–> Open Entry in Database a window containing the link to
the entry of the selected node in its original database opens. The databases are:
NCBI for genes
Monarch Initiative Explorer and OLS by EMBL-EBI for disorders (both MONDO database explorers)
Uniprot for proteins
DrugBank for drugs
Reactome for pathways
Deselect node
By right clicking on a node of interest and choosing NeDRex–> Deselect this node you can deselect the node.